Modern Computational Science - Summer School
Second Quantization: Basics, Recipes, and Applications
Martin Holthaus, University of Oldenburg
The numerical study of systems of interacting identical Bose or Fermi particles constitutes one of the basic tasks of computational science. Since such systems usually live in an extremely high-dimensional Hilbert space, they require a careful reduction of complexity before they can be treated on a computer. The language of "second quantization" provides a universal and powerful framework which enables one to formulate problems involving interacting quantum particles in a quite intuitive manner, so that useful approximations almost suggest themselves.
These two lectures will first introduce the basics of second quantization, and explore why this formalism is fairly efficient in keeping track of the (anti-)symmetry of the many-particle wave function required by Bose or Fermi statistics. Instead of proving mathematical theorems, the working principles will be illustrated with the help of famous models, such as the Bose-Hubbard model for ultracold atoms in optical lattices, the Jellium model for the interacting electron gas, and the BCS model for superconductivity. At the end, a particularly simple model will be discussed which can be programmed by the students without much effort, and which allows one to address topical questions in quantum dynamics.
Hartree-Fock self-consistent field (SCF)
Wim Klopper, Karlsruhe Institute of Technology
The lectures will be concerned with basic tools and techniques of rigorous molecular electronic structure theory, fundamental for the treatment of molecular properties. We will start with the molecular electronic Schrödinger equation and then introduce Slater determinants and the Hartree-Fock or self-consistent field (SCF) approximation. Concepts of closed- and open-shell states, canonical and localized molecular orbitals (MOs) as well as spin orbitals will be discussed in the framework of restricted and unrestricted SCF procedures. We will introduce a linear combination of atomic orbitals (LCAO) to expand the MOs leading to the Roothaan-Hall equations, and one of the lectures will address techniques for the evaluation of integrals over Gaussian functions and direct SCF procedures.
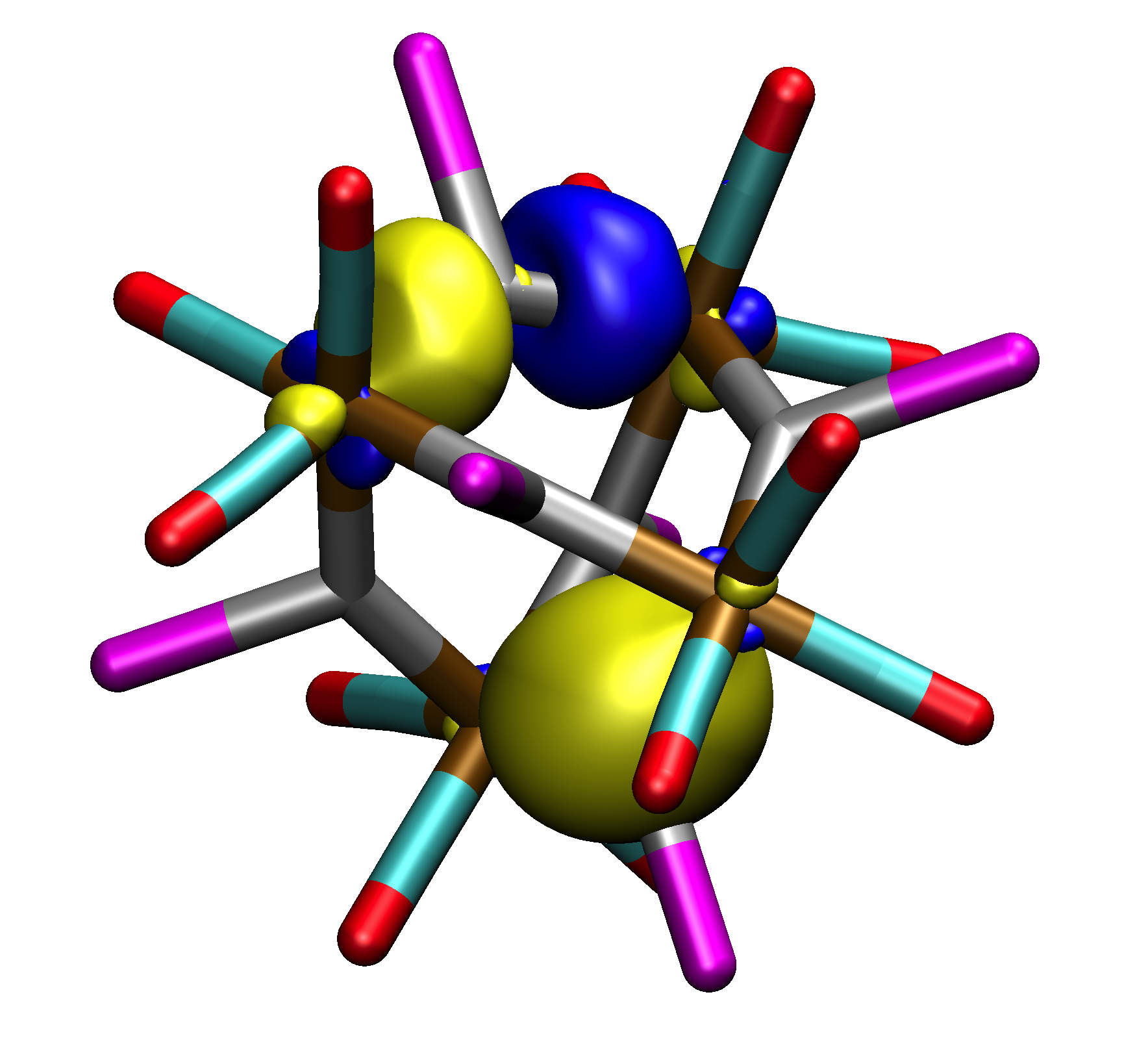
Pipek-Mezey (top) and Boys (bottom) localized molecular orbitals in the dication [{Fe(CO)3}4{SnI}6]2+.
The last lecture will review the use of explicitly correlated wave functions in quantum chemistry. Today, strong predictive power can be achieved by calculating the properties of molecules from electronic wave functions that carefully account for the instantaneous correlations between the electrons. By including terms that depend on the inter electronic distances, it becomes possible to accurately model the holes in the wave function about each electron. Functions of this type are termed explicitly corre lated wave functions.
- References
- Christof Hättig, Wim Klopper, Andreas Köhn, and David P. Tew: "Explicitly Correlated Electrons in Molecules"; Chemical Reviews; 2012; 112 (1), 4-74 [link to journal]
Advanced Quantum-Chemical Methods
VoLker Staemmler, Ruhr-University Bochum
In these lectures we will introduce advanced quantum-chemical methods that enable an accurate description of the properties of small and medium-sized molecules as well as of certain local properties of solids. In the first part we present methods going beyond the Hartree-Fock (SCF) approximation, i.e. treating the correlation problem. Several of such methods are currently widely and very successfully used for calculating properties such as energies, equilibrium geometries or potential energy surfaces for closed-shell ground states of molecular systems: Perturbation theory (MP2), configuration interaction (CI) and coupled cluster theory (CC). The characteristic features, possibilities and limitations of these approaches are discussed in some detail. A short description of density functional theory (DFT) and its relation to wave-function based methods is also given. In the second part we extend this discussion to open-shell systems and in particular to excited states, an accurate theoretical treatment of which is much more complicated than that of closed-shell states. We will describe mainly multi-reference based methods, characterized by the acronyms like CAS-SCF, CAS-CI, CAS-PT2 and MR-CI, MR-CC and show their performance using a few selected examples. A short discussion of our own approximate MR-CC approach will also be included.
In the third part some applications to solid state problems will be discussed. By representing a macroscopic solid by a finite cluster of atoms one can use the methods originally developed for molecules to quite accurately describe certain local properties of the solid. This applies in particular to insulating materials like metal oxides for which different from metals the electronic wave functions can be quite well localized. We discuss three examples from our own work: Adsorption of small molecules on metal oxide surfaces, X-ray photoemission spectra of MgO, and antiferromagnetic coupling in metal oxides. For these local properties the cluster approach using quantum-chemical methods is superior to conventional band structure methods as used in solid state physics.
The Scanning Tunneling Microscope:
A Spectroscopic Toolbox for Surface Science Research
Niklas Nilius, University of Oldenburg
Over the past 25 years, the scanning tunneling microscope (STM) has developed into an irreplaceable instrument for the characterization of solid surfaces at the atomic scale. In contrast to various indirect techniques, it provides real-space insight into atomic landscapes, including surface morphologies, defect structures, growth topologies and molecular adsorption. For a long time, the structural characterization of surfaces was the dominant field of use for this technique. However, the STM is much more than an advanced imaging tool, and offers a fascinating variety of spectroscopic approaches to probe electronic, vibrational, magnetic and even optical properties of surfaces with nanometer spatial resolution.
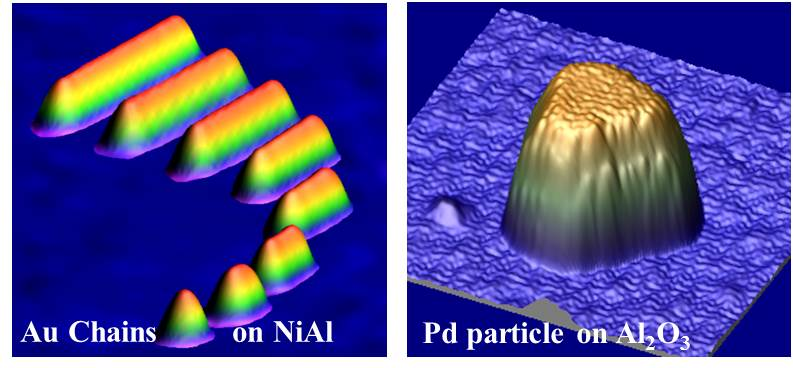
Examples for the application of the scanning tunneling microscope.
In my lecture, I provide a short introduction into the theoretical background of tunneling microscopy and the basic construction principles of the method. My focus lies however in a detailed discussion of the various spectroscopic approaches. I will demonstrate how conductance spectroscopy with the STM can be used to probe the discrete electronic states of single atoms and molecules. By aggregating individual entities into clusters, these atomic orbitals successively split into sub-states and merge into a continuous electronic band. Finally, delocalized electronic states can be identified on extended surfaces and investigated with high spatial resolution.
Inelastic tunneling spectroscopy, which monitors the energy loss of tunneling electrons on their way between tip and sample, offers another fascinating source of information. With this approach, vibrational excitations of molecules and atomic assemblies become accessible to the experiment. The method provides insight into the chemical identity of single molecules and allows one to follow single-molecule reactions. At higher loss energies, the energy transfer from tunneling electrons may even stimulate local optical excitations on the surface. Luminescence spectroscopy with the STM was consequently used to detect HOMO-LUMO excitations in organic molecules, exciton modes in semiconductor quantum-dots and plasmonic excitations in metal particles.
It is this combination of powerful imaging and spectroscopic capabilities which makes the STM to such a versatile surface science instrument.
Stochastic optimization
Alexander K. Hartmann, University of Oldenburg
Optimization algorithms lie at the heart of many problems in pure and applied Science. Well-known examples are the electronic structure of molecules and compounds, which are the focus of the present school. Other examples are the optimization of energy grids and various networks, the design of machine parts and workpieces, or the three-dimensional geometric shape of complex bio molecules, to name just a few. Advanced and powerful optimization techniques open up new possibilities for numerically tackling these and many other problems. Conversely, a deeper understanding of natural systems often leads to the development of new optimization methods (like, e.g., genetic algorithms) which may subsequently prove to be extremely useful in totally different fields.
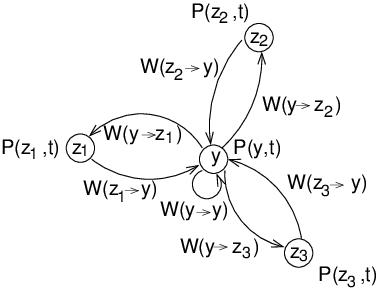
The pictures shows a small Markov chain, which is the theoretical
model on which most of the methods are based which are presented here.
Here a basic introduction to the field is given. The focus is on stochastic optimization algorithms which are very general. This means that they are usually not able to find a provable exact global optimum of a problem, nevertheless in practice they are quite powerful and able to treat much larger problems compared to exact approaches. The algorithms covered here are Monte Carlo simulations, simulated annealing, Metropolis-coupled Markov-chain Monte Carlo simulations (also known as parallel tempering), restart algorithms and genetic algorithms.
Surface Photochemistry from First Principles
Thorsten Klüner, University of Oldenburg
In this lecture, we will review the basic methodology necessary for a
faithful atomistic description of surface photochemistry with a focus
on photodesorption of diatomic molecules from surfaces. Despite its
apparent simplicity, a microscopic understanding beyond a qualitative
picture still poses a true challenge for theory. While the dynamics
of nuclear motion can be treated on various levels of sophistication,
all approaches suffer from the lack of sufficiently accurate
potential energy surfaces, in particular for electronically excited
states involved in the desorption scenario.
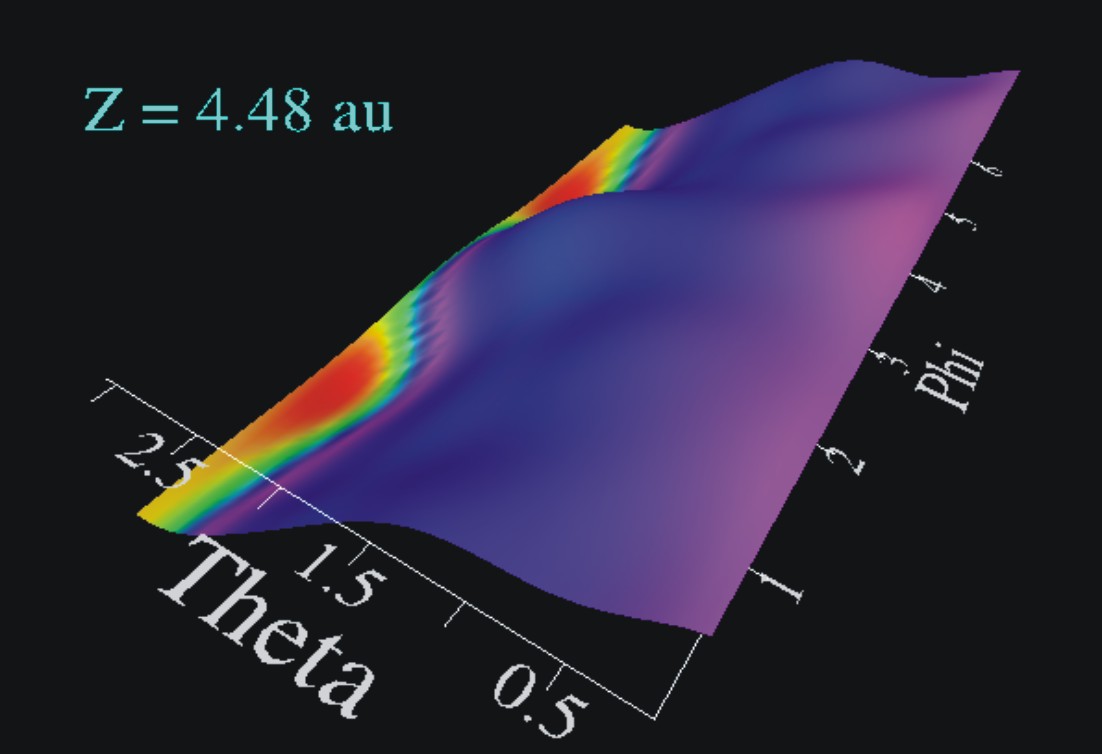
Electronically excited state of CO-molecules adsorbed on a Chromiumoxide surface.
In the last decade, we have developed a systematic and accurate methodology to reliably calculate accurate ground and excited state potential energy surfaces (PES) for different adsorbate-substrate systems. These potential energy surfaces serve as a prerequisite for subsequent quantum dynamical wave packet calculations, which allow for a direct simulation of experimentally observable quantities such as velocity distributions.
We will discuss methods to solve the time-dependent Schrödinger equation for the nuclei under the influence of a given potential energy surface. In particular, we will discuss foundations and numerical aspects of wave packet calculations including external laser fields, quantum dissipation, and optimal control.
Parallelization Strategies
Rolf Rabenseifner, High Performance Computing Center Stuttgart (HLRS)
On Sep 1-3 (Mon-Wed), the focus is on the programming models MPI, OpenMP, and PETSc. On clusters and distributed memory architectures, parallel programming with the Message Passing Interface (MPI) is the dominating parallel programming model.
The course teaches the current standard MPI-3.0, which includes library calls for blocking and non-blocking message passing, collective operations, and one-sided operations on Clusters. Additionally, MPI-3.0 offers a new one-sided shared memory model for the nodes of a cluster.
OpenMP is the major shared memory parallel programming model. Here, parallelism is expressed with directives inserted into the originally sequential source code. OpenMP offers an incremental approach to parallelism.
The PETSc library provides parallel iterative solvers for linear and non-linear equation systems. PETsc is also an interesting example for parallel software design.
Hands-on sessions (in C and Fortran) will allow users to immediately test and understand the basic constructs of MPI and OpenMP.
This part of the course is organized in collaboration with HLRS. A preliminary agenda can be found here.
Code Optimization
Georg Hager, Regionales Rechenzentrum Erlangen (RRZE)
On Sep 4-5 (Thu/Fr) the focus is on performance modeling and performance optimizations on the core and on the compute node level.
This part of the course teaches performance engineering approaches on the compute node level. This is more than employing tools to identify hotspots and bottlenecks. It is about developing a thorough understanding of the interactions between software and hardware. This process must start at the core, socket, and node level, where the code gets executed that does the actual computational work. Once the architectural requirements of a code are understood and correlated with performance measurements, the potential benefit of optimizations can often be predicted. We introduce a "holistic" node-level performance engineering strategy and apply it to different algorithms from computational science.
A preliminary agenda can be found here.
- References
- G. Hager and G. Wellein: Introduction to High Performance Computing for Scientists and Engineers. CRC Press, ISBN 978-1439811924, 356 pages, July 2010